空间转录组测序|Cell-小鼠肠绒毛的带状分布模式


肠上皮主要由单层上皮细胞组成,具有高度折叠结构【1】。每天,肠隐窝(crypt)中成千上百个新生肠上皮细胞沿着肠壁内小小的、手指似的凸起——绒毛(villus)轴向移动,仅3-5天后,这些肠上皮细胞就从绒毛顶端处脱落,掉入肠腔内【2】。

小肠上皮细胞结构示意图【3】
虽然前人研究结果揭示了沿小肠隐窝-绒毛轴向出现的转录组变化,但bulk测序难以检测到沿绒毛方向出现的空间表达变化和肠上皮细胞的异质性【4-6】。
此外,近期使用单细胞RNA测序(scRNA-seq)技术确定了肠内细胞的异质性,然而在分离单个细胞过程中丢失了其空间信息,导致这些研究无法解答绒毛的空间异质性和特异性【7-11】。
为此,Weizmann科学研究所的研究团队使用激光捕获显微切割(LCM)技术获取绒毛特定位置处的细胞样本进行RNA-seq,以得到不同位置处的标志基因。根据这些标志基因,研究者通过重新分析他人scRNA-seq数据,确定了数据中单个细胞的位置,揭示了肠上皮细胞状态的空间特异性。
这一研究成果在2018年发表于Cell。

影响因子:36.216
第一作者:Weizmann科学研究所Andreas E. Moor教授
通讯作者:Weizmann科学研究所Andreas E. Moor教授、Shalev Itzkovitz教授
通讯作者实验室:Shalev Itzkovitz教授实验室
开放获取数据:bulk测序数据存储于NCBI,编号GSE109413;重建空间结构的算法脚本存储于Github;脚本对应的原始和中间输入文件存储于Zenodo,包括单细胞数据Seurat分析输出的相关文件。

研究者从3个8周大雄性C57BL/6小鼠的空肠近端取得了组织块,将冷冻组织块切成8um厚度的切片。随后,使用脉冲UV激光切割染色后干燥切片上的组织。每个老鼠空肠切片中,8-10个长度超过500um的绒毛被用于显微切割。每个绒毛上皮按照长度等分成5份进行显微切割(图1),并进行RNA-seq。

图1 使用LCM获取5等分绒毛样本以进行RNA-seq【2】。

为了取得肠上皮细胞的标志基因集,研究者使用LCM分离得到小鼠空肠中绒毛底-顶端间5等分的上皮细胞样本,并进行RNA-seq,据此确定了62个绒毛底端标志基因以及43个绒毛顶端标志基因(图2a)。
根据顶端和底端标志基因间总表达水平的比值(图2b),scRNA-seq数据集【8】中除杯状细胞和肠内分泌细胞以外,每个细胞被赋予了一个无单位空间坐标x(图2c)。随后,研究者计算5个LCM位置处RNA-seq数据的顶端和底端标志基因总表达水平比值x’。通过比较x与x’,确定scRNA-seq数据集中单个细胞的空间位置。

a

b

c
图2 构建的绒毛肠上皮细胞的空间表达谱【2】。a是绒毛底端标志基因(左)和绒毛顶端标志基因(右)表达水平图谱;b是前人scRNA-seq数据集中,除杯状细胞和肠内分泌细胞以外,单个细胞的底端标志基因的总表达水平(左)、顶端标志基因的总表达水平(中)和表达水平比值(右);c是scRNA-seq数据集中成熟肠上皮细胞和祖细胞群的t-SNE降维聚类结果,颜色代表着每个细胞在绒毛中的参考位置。
为了验证构建空间图谱的准确性,研究者使用单分子荧光原位杂交(smFISH)验证15个肠上皮基因的表达模式,结果证实了空间图谱的准确性(图3a)。此外,随机选择50个标志基因构建的空间图谱的标准误较小,但随机选择20个标志基因构建的空间图谱的标准误较大(图3b)。
综上,研究者使用的这一方法能够可靠的重建肠绒毛的带状表达模式。

a

b
图3 a是使用smFISH确定了重建的带状图谱的准确性:深蓝色线表示的是scRNA-seq数据中平均表达水平;淡蓝色区域表示的是scRNA-seq均值标准误;深红色线表示的是smFISH中平均表达水平,淡红色区域表示的是smFISH均值标准误;b是随机使用少量标志基因构建空间图谱的均方误差(左)以及使用不同数量标志基因重建的带状图谱中,部分基因的表达水平(右)【2】。

为了研究绒毛带状分布模式背后的分子机制,研究者将scRNA-seq数据按照绒毛底端-顶端的顺序,聚类成5个不同的基因聚群,并进行富集分析。结果显示(图4):
❖ 聚群1包含从肠隐窝到绒毛方向,表达逐渐降低的基因。这种降低还体现在翻译、转录及RNA剪切水平的整体减少。这似乎意味着随肠上皮细胞沿绒毛轴向移动,其生物合成能力逐渐下降;
❖ 聚群2富集了线粒体功能相关基因,这可能是对氧浓度逐渐降低的适应【12】。此外,该聚群还包含了谷胱甘肽转移酶活性,以及在绒毛底端高表达但在临近肠隐窝处不表达的急性时相反应基因;
❖ 聚群3富集的肠转运功能在绒毛中部有峰值;
❖ 在绒毛中部前,聚群4中的基因表达逐渐增加,其中包含许多绒毛刷状缘组分;
❖ 聚群5包含脂蛋白生物合成、细胞粘附程序相关基因以及旁斑和斑的IncRNA(长链非编码RNA)标记,这些都沿绒毛顶端呈单调递增。
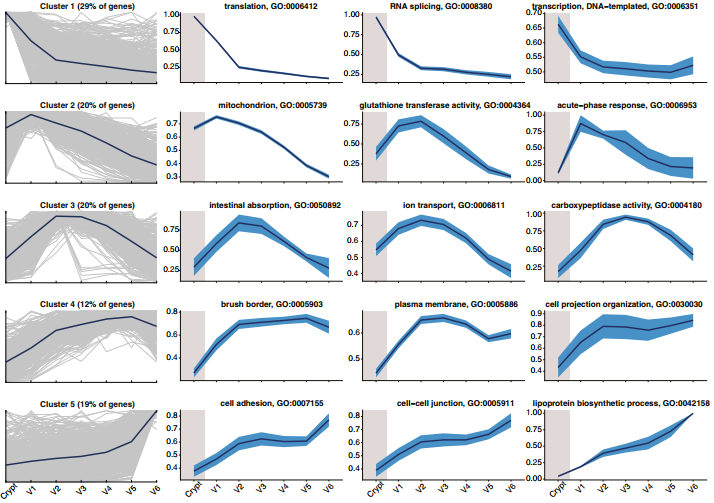
图4 绒毛肠上皮细胞的部分富集分析结果【2】。其中,最左侧一列中深蓝色线表示的是基因聚群的表达均值,灰色线代表着每个基因的表达水平。第2-第4列中,深蓝色线表示的是富集相关基因的平均表达水平,淡蓝色区域表示的是均值的标准误。

在使用前人scRNA-seq数据重建的带状空间表达图谱中,研究者发现:
❖ 绒毛底端部分区域中肠上皮细胞明显表达Reg家族成员【13-15】,以及其他参与微生物-宿主相互作用的肽,如Nlrp6【16】、Il18【17】、Ccl25【18】和抗菌Lypd8【19】(图5a);
❖ 与传统无菌小鼠RNA-seq数据相比【20】,Reg3b和Reg3g是表达减少最明显的两个基因(图5b);
❖ 使用抗生素移除微生物后【21】,Reg3b和Reg3g表达明显降低(图5c);
❖ smFISH结果显示,与对照组相比,无菌小鼠的肠绒毛底端Reg3g表达显著降低(图5d)。
Reg家族基因属于钙依赖的凝集素基因并编码小的分泌蛋白。这些发现表明,肠隐窝的上部存在一个抗菌区域。该区域可能作为肠隐窝干细胞的“守门人”,尽可能避免其与致病微生物接触。

a

b

c

d
图5 绒毛底端细胞表达依赖微生物的抗菌程序【2】。a是参与微生物-宿主相互作用基因的带状表达热图;b是前人研究中,同窝出生小鼠在常规或无菌环境下喂养2周后,mRNA表达水平火山图(左)和Reg3b、Reg3g的表达水平(右)【20】;c是前人研究中,对照小鼠以及抗生素处理30天的小鼠Reg3b和Reg3g表达水平【21】;d是常规或无菌环境下喂养的小鼠,其肠隐窝和临近绒毛底端区域处Reg3g表达的smFISH分析结果(左)以及Reg3g的mFISH染色定量结果(右)。

肠上皮细胞吸收多种营养物质,包括糖类、氨基酸以及脂质。为了研究营养转运体的空间分布模式,研究者分析了scRNA-seq数据中、与不同类型营养物质吸收有关的基因的表达水平模式,发现(图6):
❖ 关键营养物质家族的转运体表现出不同的带状分布模式;
❖ 绒毛中部富集了氨基酸和糖类转运体,编码主要肽链转运体Pept1的Slc15a1基因表达水平在肠绒毛底端处发生转变;
❖ 在绒毛顶端,乳糜微粒形成所必须的胆固醇转运体Npc1l1以及脂蛋白合成机制相关基因的表达达到峰值,这解释了前人的研究发现,即脂质灌胃后,很快在绒毛顶端处观察到更高的乳糜微粒浓度【22】;
这些结果似乎意味着肠上皮细胞在绒毛轴向不同位置处,倾向吸收的营养物质不同。

图6 选定基因的热图和平均带状图谱,这些基因对不同类型营养物质吸收十分重要【2】。

研究者使用前人的scRNA-seq数据重建了空间图谱,发现绒毛顶端处不同信号通路和转录信号基因集表达水平的显著增加。进一步分析后,发现:
❖ 绒毛顶端处Egfr表达增加可能启动了细胞粘着性的改变【24】,这对随后的细胞脱落十分重要(图7a);
❖ 绒毛顶端细胞的嘌呤降解基因信号表达增加,且Nt5e蛋白质及多数外核苷酸酶位于绒毛顶端的内腔,意味着肠具有抗炎性功能【24】。这些信号程序可能对避免过度地免疫响应(针对肠道中的微生物)十分重要(图7b)。

a

b
图7 重建的空间结构揭示了肠绒毛顶端的表达程序。a是绒毛顶端处的信号和转录程序,其中深蓝色线是基因平均表达水平,浅蓝色区域是均值的标准误;b是绒毛顶端处嘌呤降解标记基因表达水平t-SNE图(左上),Ada转录的smFISH结果(左下)以及绒毛顶端处嘌呤降解基因的互作模型(右)。
肠上皮细胞是沿绒毛移动、仅存活几天的短寿命细胞,接近许多蛋白质的典型半衰期【25】。因而研究者认为,mRNA总量可能不存在沿着绒毛轴向逐渐降低,以减少肠上皮细胞内蛋白质的含量。
为了评估绒毛不同高度处的肠上皮细胞蛋白质组,研究者根据Nt5e的表达划分出3个肠上皮细胞聚群并进行蛋白质组学质谱研究和RNA-seq数据验证,发现根据Nt5e表达划分出的3个肠上皮细胞聚群与根据转录组构建的空间表达带状图谱较为相似(图8)。

图8 使用带状重建算法推断的每个细胞聚群(根据Nt5e表达划分出的3个肠上皮细胞聚群)的空间坐标【2】。
此外,从Nt5e表达水平中等聚群到Nt5e表达水平高的聚群,载脂蛋白表达降低(图9)。乳糜微粒中的载脂蛋白与脂质一同分泌,因而更高水平的mRNA、更低的细胞内蛋白质水平与绒毛顶端处脂质分泌增加相一致【22】。

图9 根据Nt5e表达划分的3个肠上皮细胞聚群的蛋白质丰富度。

为了阐述肠上皮细胞沿绒毛轴向迁移时细胞状态的连续转换,研究者使用谱系追踪法跟踪Lgr5+干细胞的克隆子代,发现这些克隆细胞有着连续转变,从绒毛底端Reg1、绒毛中部Slc2a2到绒毛顶端基因Ada(图10a)。
当对前人的scRNA-seq数据进行伪时间分析时,这种逐渐转变也很明显。进一步证实了肠上皮细胞沿绒毛轴向迁移时,其细胞状态按照一定顺序发生连续转变(图10b、c)。

a

b
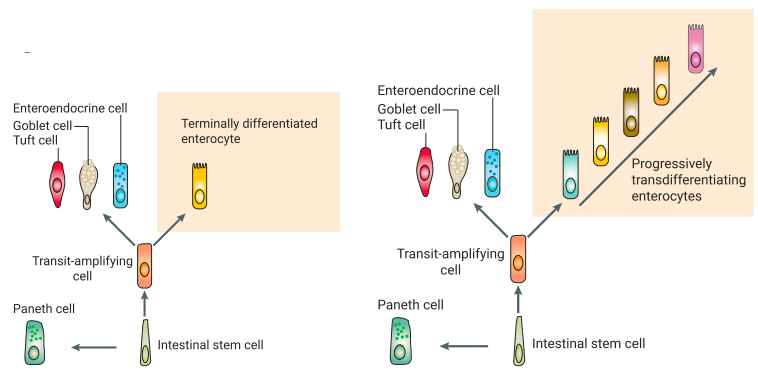
c
图10 肠上皮细胞沿绒毛轴向移动时的转分化。a是Lgr5+干细胞的谱系追踪结果;b是肠上皮细胞的伪时间轨迹分析结果;c中左图是肠上皮谱系经典观点示意图,认为只有一种类型的肠上皮细胞,右图是研究者揭示肠上皮细胞沿绒毛轴向移动时,细胞状态发生连续转变。

揭示了小肠上皮细胞广泛的空间异质性:绝大多数基因表达具有明显的带状分布模式,几乎没有基因沿绒毛轴向具有一致的表达水平。肠上皮细胞不是终末分化细胞,而是在沿绒毛轴向移动过程中发生连续的转分化。
肠隐窝处的干细胞小生境似乎受到绒毛底端的一层肠上皮细胞的保护,这些细胞表达炎性小体成分并分泌抗微生物Reg蛋白。这些肠上皮细胞可能辅助Paneth细胞保护肠隐窝处驻留的干细胞。
绒毛不同区域处有着不同的特定营养物质吸收机制,可能与更高效的营养吸收有关。
绒毛顶端细胞似乎负责指挥免疫模块程序,可能对宿主-微生物间相互作用有着重要意义。
结合证据来看,研究者提出一种假设:绒毛内腔不同空间位置处的营养物质浓度不同,这种空间异质性导致带状分布的微生物小生境,即不同的微生物倾向定殖于绒毛的不同位置。

每天,肠壁内绒毛底部产生了成百上千个新细胞(比其他任何组织都多),这些细胞沿着绒毛轴向迁移4天后就从绒毛顶端脱落。
以往科学家们相信一旦肠隐窝处干细胞分化成不同类型的细胞,这就是最终的分化——即,一旦干细胞成熟为,例如肠上皮细胞这一肠壁内最常见的细胞类型,它的分化就已经结束了。但是Weizmann的科学家展示了当肠上皮细胞沿着绒毛迁移时,它们发生持续分化,在不同位置实现不同功能。
“它们仍被称为肠上皮细胞,但我们发现不同亚型具有如此不同的特征。实际上,它们应是不同类型的细胞,”Itzkovitz说到。
该研究团队开发的这一方法可用于构建身体不同组织以及肿瘤的详细细胞图谱。他们也为研究疾病发生发展提供了新的可用手段,例如,检查小肠内壁不符合细胞位置的表达基因是否参与了炎症性肠病。
2.Moor, Andreas E., et al. (2018). "Spatial reconstruction of single enterocytes uncovers broad zonation along the intestinal villus axis." Cell 175(4): 1156-1167.
3.Circu, Magdalena L., and Tak Yee Aw. (2011). "Redox biology of the intestine." Free radical research 45(11-12): 1245-1266.
4.Mariadason, John M., et al. (2005). "Gene expression profiling of intestinal epithelial cell maturation along the crypt-villus axis." Gastroenterology 128(4): 1081-1088.
5.Stegmann, Anders, et al. (2006). "Metabolome, transcriptome, and bioinformatic cis-element analyses point to HNF-4 as a central regulator of gene expression during enterocyte differentiation." Physiological genomics 27(2): 141-155.
6.George, Michael D., et al. (2008). "In vivo gene expression profiling of human intestinal epithelial cells: analysis by laser microdissection of formalin fixed tissues." BMC genomics 9(1): 209.
7.Grün, Dominic, et al. (2015). "Single-cell messenger RNA sequencing reveals rare intestinal cell types." Nature 525(7568): 251.
8.Yan, Kelley S., et al. (2017). "Intestinal enteroendocrine lineage cells possess homeostatic and injury-inducible stem cell activity." Cell stem cell 21(1): 78-90.
9.Haber, Adam L., et al. (2017). "A single-cell survey of the small intestinal epithelium." Nature 551(7680): 333.
10.Herring, Charles A., et al. (2018). "Unsupervised trajectory analysis of single-cell RNA-seq and imaging data reveals alternative tuft cell origins in the gut." Cell systems 6(1): 37-51.
11.Glass, Leslie L., et al. (2017). "Single-cell RNA-sequencing reveals a distinct population of proglucagon-expressing cells specific to the mouse upper small intestine." Molecular metabolism 6(10): 1296-1303.
12.Zheng, Leon, Caleb J. Kelly, and Sean P. Colgan. (2015). "Physiologic hypoxia and oxygen homeostasis in the healthy intestine. A review in the theme: cellular responses to hypoxia." American Journal of Physiology-Cell Physiology 309(6): C350-C360.
13.Vaishnava, Shipra, et al. (2011). "The antibacterial lectin RegIIIγ promotes the spatial segregation of microbiota and host in the intestine." Science 334(6053): 255-258.
14.Burger-van Paassen, Nanda, et al. (2012). "Mucin Muc2 deficiency and weaning influences the expression of the innate defense genes Reg3β, Reg3γ and angiogenin-4." PloS one 7(6): e38798.
15.Earle, Kristen A., et al. (2015). "Quantitative imaging of gut microbiota spatial organization." Cell host & microbe 18(4): 478-488.
更多信息请关注我们的网站
www.geno-truth.com/singlecell
联系电话:4000-672-118
服务邮箱:service@geno-truth.com

●文献解析|Science—scRNA-seq揭示水螅再生机制
文献解析|Nature—单细胞RNA测序首次揭秘人类胎肝造血作用(上)
文献解析|Nature—单细胞RNA测序首次揭秘人类胎肝造血作用(下)
单细胞空间转录组测序|Cell—小鼠原肠胚形成中期RNA空间模式





